标签:
转自:http://www.gogoqq.com/ASPX/8390905/JournalContent/1303140588.aspx
研究了近半年的算法,记录下来给自己一个交代,也应该是考G前地最后一篇日志了。
Weighted Gene Co-Expression Network Analysis中文名有翻译成加权关联网络分析的,感觉不是很恰当,英文来得比较直接。本来是佟昊从老汪那拿的一个课题,因为看起来比较有意思就把文章找来慢慢啃,到现在算是捣鼓出点名堂了。方法是UCLA的一个教授提出来的,在文章中他将其归类到系统生物学的研究方法中,不过个人认为由于其分析水平还是只停留在DNA芯片上,并未到达系统的程度,但是方法本身还是能够预见一些incisive idea的。我打算先介绍方法的基本思想,然后把联系网上公布的sample data将该算法完整实现一遍,并解释一些自己在看的时候遇到的关键问题。
Weighted Gene Co-Expression Network Analysis(以下简称WGCNA),是一种从芯片数据中挖掘模块(module)信息的算法。在该方法中module被定义为一组具有类似表达谱的基因,如果某些基因在一个生理过程或不同组织中总是具有相类似的表达变化,那么我们有理由认为这些基因在功能上是相关的,可以把他们定义为一个模块(module)。这似乎有点类似于进行聚类分析所得到结果,但不同的是,WGCNA的聚类准则具有生物学意义,而非常规的聚类方法(如利用数据间的几何距离),因此该方法所得出的结果具有更高的可信度。当基因module被定义出来后,我们可以利用这些结果做很多进一步的工作,如关联性状(随后会以这方面的应用为Example),代谢通路建模,建立基因互作网络,甚至进行eQTL(这个确实很方便,不过前提是实验题有钱去杂那么多芯片)。不过我个人从中获益最多的是能加深人们对于生物体所选择的这种Scale-Free Topology网络调控的思考(下文会提到)。
WGCNA所分析的数据是芯片数据(当然需要杂很多芯片,比如若要研究细胞凋亡,那么使用改方法需要实验者提供细胞凋亡各个时期的芯片数据,以了解这一生理过程中细胞内所有基因的表达变化)。
在co-expression network中,每一个基因在一个特定时间或空间的表达情况被视做一个点(node),可以简单的理解成一张芯片上的一个基因的表达状况就是网络中的一个node。如果我们做了80张芯片,每张芯片上有8000个基因,那么我们可以用一个80*8000的矩阵来表示实验结果。为了得到基因间的关联情况,我们需要计算任何两个基因间的相关系数(文章中采用Person Coefficient),在经过该步运算以后,我们可以得到一个8000*8000的实对称阵S,sij表示第i个基因和第j个基因的Person Coefficient,即两个基因的表达谱相似性。
下一步的分析是该方法的第一个靓点。为了知道两个基因的表达谱是否具有相似性,需要人为规定一个阈值,只有当基因间的Person Coefficient达到这一阈值后(如0.8)我们才认为这两个基因是相似的,否则则不相似。为此人们定义了一个Adjacency Matrix,很显然在通过以上步骤处理S矩阵后,得到的Adjacency Matrix将是一个0/1矩阵(该矩阵主对角线上元素被规定为0,这一点很重要)。但是这种分析方法存在一个很明显的局限,即我们没有理由认为Person Coefficient为0.8的两个基因与Coefficient为0.79的两个基因是有显著差别的,但是以上算法却无法避免这一处境。而WGCNA采用了一种基于软阈值的判定方法很好地避免了这一问题。软阈值的思想是通过权函数将Adjacency Matrix中的元素连续化(所以方法才称之为Weighted Network),常用的权函数包括sigmoid函数和power函数。

那么aij同样是可分解的,这一点很好证明。拥有这一性质的优势在于可以简化计算量,当实对称阵是可以分解的时候,我们只需要利用其分解后的一个向量就可以表示该矩阵,因此在实际应用中能够简化计算机的运算内存使用。 如果直接拿Adjacency Matrix中的基因相关性指标进行module识别则未免有些过于简单了,为了保证芯片信息的充分利用,文章的作者提出了计算另一矩阵——topological overlap matrix(TOM)来衡量两个基因的相关性。建立这一矩阵的思想在于,任何两个基因的相关性不仅仅由他们的表达相似性直接决定,它还将A基因通过B基因与C基因的作用相关性纳入AC基因的TOM矩阵值中,以更精确地描述基因表达谱的类似性。

利用这种方式定义TOM矩阵中各元素的值是非常巧妙的,它很好地满足了我们所希望达到的目的。分子中对于lij
的定义,表示基因i通过任何基因与j关联的adjacency值,并将它们相加,而aij
则表示基因i与基因j间的直接关联性。分母的定义保证了wij
始终在0,1之间,我们可以考虑极端情况。当Adjacency Matrix中除主对角线元素外所有元均为1时,

上式直接看不容易看懂,把加和表达式展开之后则容易理解。
值得注意的是,WGCNA方法只考虑了一阶基因关联,更高级的关联可以用类似的方式去表示,但是并无此必要。首先芯片数据本身存在噪音,过度地提取信息未必会得到更好地结果,而且计算高阶关联会使算法的复杂度显著增大,即使高配置的服务器也未必能满足计算要求。
为方便后面的module identification,还需定义一个dissimilarity matrix。根据前人的研究,方式如下:
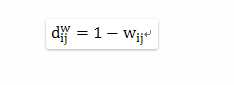
其中dij
表示dissimilarity matrix中的元,这一方程只是用1减去wij
得到dissimilarity matrix,在dij
上加指数的原因是empirical研究的结果,当使用指数形式的dissimilarity进行聚类分析会得到更distinct gene module。 得到了dissimilarity matrix后,我们所需要进行的工作就是聚类了,文章中使用的是hierarchical clustering方法,各种聚类方法的优劣不在本文的讨论范围中。使用聚类分析之后,各个module的identification也就完成了。
了解了整个分析流程,我们有必要再进一步了解一下分析细节。
首先是权函数的参数选择,由于power function有一参数
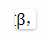
这一参数的选择势必影响着module identification的结果。
为了选择一个合适的参数值,我们有必要重新审视基因互作网络的构造。 网络的数学名称是图,在图论中对于每一个节点有一个重要概念,即:度。一个点的度是指图中该点所关联的边数。如下图,如果不加以思考,人们很容易认为生活中常见的网络会是一种random network,即每一个节点的度相对平均。然而第二种图,即scale-free network才是一种更稳定的选择。Scale-free network具有这样的特点,即存在少数节点具有明显高于一般点的度,这些点被称为hub。由少数hub与其它节点关联,最终构成整个网络。这样的网络的节点度数与具有该度数的节点个数间服从power distribution。这为我们寻找最佳参数提供了理论依据。 这里做一点扩展,我认为时非常有必要的。只要我们愿意抽象,Scale-free network大量存在于的生活中。人们的社交网络、生物基因蛋白质的相互作用、计算机网络甚至sexually transmitted diseases均有这层关系。生物体选择scale-free network而不是random network是有它进化上的原因的,显然对于scale-free network,少数关键基因执行着主要功能,这种网络具有非常好的鲁棒性,即只要保证hub的完整性,整个生命体系的基本活动在一定刺激影响下将不会受到太大影响,而random network若受到外界刺激,其受到的伤害程度将直接与刺激强度成正比。
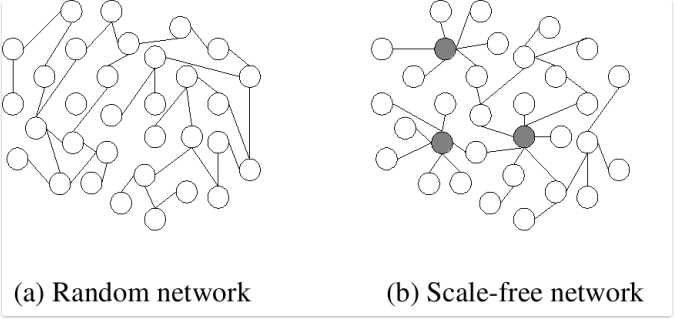
Random network (a) and scale-free network (b). In the scale-free network, the larger hubs are highlighted. 图片来源:http://en.wikipedia.org/wiki/File:Scale-free_network_sample.png有了这一理论基础,我们可以尝试一系列权函数的参数值,如
然后找出得到的网络最符合scale-free network的frequency distribution的这样一个
作为后续分析使用。但在实际寻参过程中,存在一个trade-off,即在最大化topology free network的回归系数R2
与保证节点的关联数目上为此消彼长的关系,这其实可以建立一个优化模型,但是文章的作者没有继续深入研究得到客观的寻参标准。尽管作者已经建立了一个scale-free topology criterion,但分析过程依旧有很大的主观成分。 Post-module analysis 当建立完module后,为了方便处理module与其它数据的相关性,如性状信息,有必要在每一个模块中定义一个特征基因,这一基因能在可接受的信息损失程度下代表这一module的特征,这样做的一个很大好处是能简化计算,即使在数据量极为庞大的时候也能快速地得到结果。 在后续的分析中,作者还比较了hard/soft-threshold方法建立网络的clustering coefficient的相关性质,以及它们对网络connectivity的影响,作者这样分析是为了说明soft-threshold方法相比hard-threshold的优势,由于涉及到较深的图论方面的知识,并且与module的建立无关,因此不在此处进一步讨论。 参考文献: Bin Zhang, Steve Horvath, A General Framework for Weighted Gene Co-Expression Network Analysis, Statistical Applications in Genetics and Molecular Biology, Departments of Human Genetics and Biostatistics, University of California at Los Angeles, 2005, Volume 4, issue 1, Article 17.
标签:
原文地址:http://www.cnblogs.com/Acceptyly/p/4391208.html