标签:code 构建 最小二乘法 ide read ado cluster pair log
相关链接
http://journals.sagepub.com/doi/10.3181/0903-MR-94 (冠状病毒的Minireview)
http://www.biotrainee.com/thread-2253-1-1.html (系统发育树相关)
https://blog.csdn.net/Cccrush/article/details/90695891 (详细介绍进化树的几种构建方法和原理)
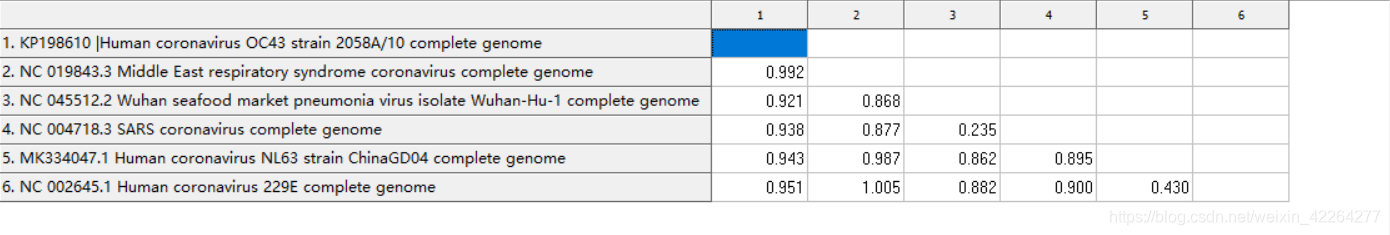
物种:冠状病毒中能够感染人的7种病毒
序列来源:NCBI上已经公布的Ref序列,我们只采用了其中的6种。


以上的4种方法其实都属于距离法,即通过计算各物种之间的进化距离来作为建树的依据。
实际上还有一类建树的法则:Character-based methods 特征法,这里先跳过去,日后在看(挖坑ing)。
上面的几种工具在EBI的网站上都有公布(实际上里面还有很多的工具可以实现多序列比对),我们采用了其中的MUSCLE方法+ClustalW方法+MAFFT方法,能够直接得到最终的建树结果。
相关网页:
https://www.ebi.ac.uk/Tools/msa/muscle/
https://www.ebi.ac.uk/Tools/msa/clustalo/
Muscle:Accurate MSA tool, especially good with proteins. Suitable for medium alignments.

ClustalW:New MSA tool that uses seeded guide trees and HMM profile-profile techniques to generate alignments. Suitable for medium-large alignments.

MAFFT:MSA tool that uses Fast Fourier Transforms. Suitable for medium-large alignments.

后面两个的结果相近,可能更加接近真实情况。
# Muscle
(
(
KP198610:0.22253,
(
NC_002645.1:0.16531,
MK334047.1:0.15607)
:0.08099)
:0.01856,
NC_019843.3:0.22538,
(
NC_045512.2:0.09935,
NC_004718.3:0.10340)
:0.11661);
# ClustalW
(
(
NC_019843.3:0.23351,
(
NC_045512.2:0.09863,
NC_004718.3:0.10330)
:0.12454)
:0.02357,
KP198610:0.23317,
(
MK334047.1:0.16005,
NC_002645.1:0.16886)
:0.09141);
# MAFFT
(
KP198610:0.23000,
(
MK334047.1:0.15815,
NC_002645.1:0.16536)
:0.08813,
(
NC_019843.3:0.22966,
(
NC_045512.2:0.09772,
NC_004718.3:0.10361)
:0.12929)
:0.03177);

标签:code 构建 最小二乘法 ide read ado cluster pair log
原文地址:https://www.cnblogs.com/zhengjm/p/12640256.html