标签:alt detail 文件类型 tor each ica size equal 通过
最近帮女朋友做毕业设计的时候用到了 PAML这个软件的codeml功能,发现网上相关的资料很少,于是把自己踩的一些坑分享一下,希望能帮到其他有相同困难的人
一、下载与安装
PAML软件下载地址
http://abacus.gene.ucl.ac.uk/software/paml4.9j.tgz
DAMBE软件下载地址
http://dambe.bio.uottawa.ca/DAMBE/dambe_install_win.aspx
二、使用方法
首先准备好你的fas文件
我们需要将fas文件转换一下格式,方法很多,我这边说两种方法,这两种方法最后得到的文件内容完全相同,只是后缀名不同
方法一:
使用python脚本转换
将你的*.fas文件与脚本放在同一目录下,执行脚本,会生成一个.phy文件
import re with open(‘seven.fas‘, ‘r‘) as fin: sequences = [(m.group(1), ‘‘.join(m.group(2).split())) for m in re.finditer(r‘(?m)^>([^ \n]+)[^\n]*([^>]*)‘, fin.read())] with open(‘seven.phy‘, ‘w‘) as fout: fout.write(‘%d %d\n‘ % (len(sequences), len(sequences[0][1]))) for item in sequences: fout.write(‘%-20s %s\n‘ % item)
方法二:
使用DAMBE软件转换格式
1.打开DAMBE,选择 File -> Open standard sequence file -> 文件类型选择为包含 fas 类型 -> 选择你的fas文件
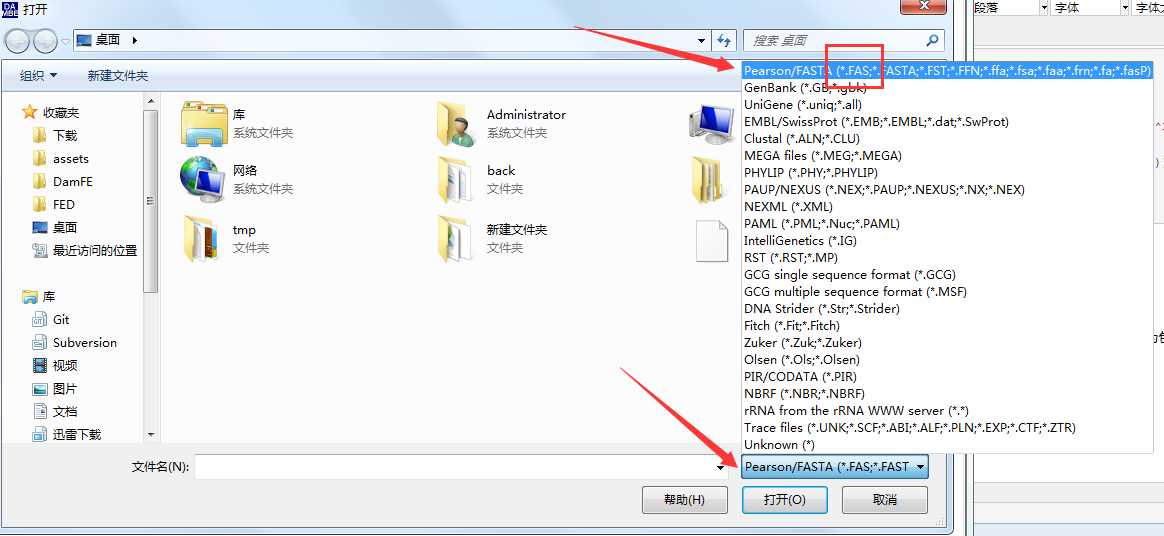
2.点击 go
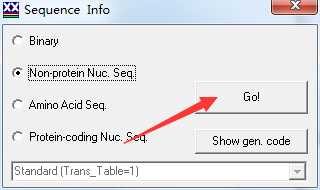
3.点击 File -> save or convert sequence format -> 选择 paml 格式
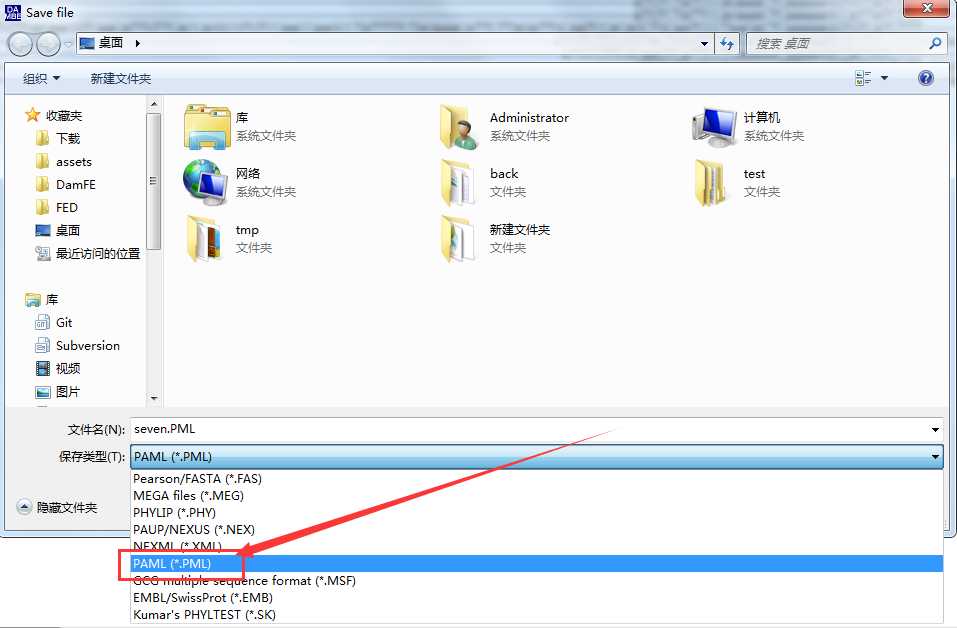
4.手动修改 *.pml 的后缀名为 *.nuc
通过以上两个方法会得到一份 *.phy 或者 *.nuc 文件
接下来需要去除序列中的终止密码子
你可以全选文件内容查找替换 将 TAG/TAA/TGA 替换为 ---
也可以使用下面这个python脚本
import re with open(r‘seven.phy‘, ‘r‘) as f: content = f.read() content = content.replace("TAG","---") content = content.replace("TAA", "---") content = content.replace("TGA", "---") # print(content) with open(‘sevenend.phy‘, ‘w‘) as f: f.write(content)
会生成一个去除过终止密码子的文件
现在将这个处理过后的序列文件*.phy与树文件、配置文件codeml.ctl三个放在 \paml4.9j\bin 目录下
配置文件codeml.ctl内容如下可参考 一般修改前面三行即可 按顺序为序列文件名 树文件名 输出文件名
seqfile = seven.nuc * sequence data filename treefile = Newick * tree structure file name outfile = test.txt * main result file name noisy = 0 * 0,1,2,3,9: how much rubbish on the screen verbose = 0 * 0: concise; 1: detailed, 2: too much runmode = -2 * 0: user tree; 1: semi-automatic; 2: automatic * 3: StepwiseAddition; (4,5):PerturbationNNI; -2: pairwise seqtype = 1 * 1:codons; 2:AAs; 3:codons-->AAs CodonFreq = 2 * 0:1/61 each, 1:F1X4, 2:F3X4, 3:codon table * ndata = 5504 clock = 0 * 0:no clock, 1:clock; 2:local clock; 3:CombinedAnalysis aaDist = 0 * 0:equal, +:geometric; -:linear, 1-6:G1974,Miyata,c,p,v,a aaRatefile = dat/jones.dat * only used for aa seqs with model=empirical(_F) * dayhoff.dat, jones.dat, wag.dat, mtmam.dat, or your own model = 0 * models for codons: * 0:one, 1:b, 2:2 or more dN/dS ratios for branches * models for AAs or codon-translated AAs: * 0:poisson, 1:proportional, 2:Empirical, 3:Empirical+F * 6:FromCodon, 7:AAClasses, 8:REVaa_0, 9:REVaa(nr=189) NSsites = 0 * 0:one w;1:neutral;2:selection; 3:discrete;4:freqs; * 5:gamma;6:2gamma;7:beta;8:beta&w;9:betaγ * 10:beta&gamma+1; 11:beta&normal>1; 12:0&2normal>1; * 13:3normal>0 icode = 0 * 0:universal code; 1:mammalian mt; 2-10:see below Mgene = 0 * codon: 0:rates, 1:separate; 2:diff pi, 3:diff kapa, 4:all diff * AA: 0:rates, 1:separate fix_kappa = 0 * 1: kappa fixed, 0: kappa to be estimated kappa = 2 * initial or fixed kappa fix_omega = 0 * 1: omega or omega_1 fixed, 0: estimate omega = .4 * initial or fixed omega, for codons or codon-based AAs fix_alpha = 1 * 0: estimate gamma shape parameter; 1: fix it at alpha alpha = 0. * initial or fixed alpha, 0:infinity (constant rate) Malpha = 0 * different alphas for genes ncatG = 8 * # of categories in dG of NSsites models getSE = 0 * 0: don‘t want them, 1: want S.E.s of estimates RateAncestor = 1 * (0,1,2): rates (alpha>0) or ancestral states (1 or 2) Small_Diff = .5e-6 cleandata = 1 * remove sites with ambiguity data (1:yes, 0:no)? * fix_blength = 1 * 0: ignore, -1: random, 1: initial, 2: fixed, 3: proportional method = 0 * Optimization method 0: simultaneous; 1: one branch a time * Genetic codes: 0:universal, 1:mammalian mt., 2:yeast mt., 3:mold mt., * 4: invertebrate mt., 5: ciliate nuclear, 6: echinoderm mt., * 7: euplotid mt., 8: alternative yeast nu. 9: ascidian mt., * 10: blepharisma nu. * These codes correspond to transl_table 1 to 11 of GENEBANK.
在此目录下打开命令行
输入一下命令即可
codeml
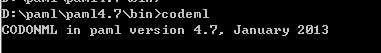
当前目录下就会出现结果文件 test.txt 以及其他文件了
在过程中我遇到过许多报错提供给大家参考一下
67 columns are converted into ??? because of stop codons
这个报错是因为没有去除文件中的终止密码子,可以参考上面的步骤去除
Error: Error in sequence data file: . in 1st seq.?.
Error: check #seqs and tree: perhaps too many ‘(‘?.
Make sure to separate the sequence from its name by 2 or more spaces.
以上报错均为你的序列文件内容/格式有问题,麻烦按照上面的步骤重新生成序列文件或者参考其他人的文件格式
标签:alt detail 文件类型 tor each ica size equal 通过
原文地址:https://www.cnblogs.com/chestnut-egg/p/12894987.html