标签:rar 之间 关联性 als 关联 包括 syn 输出 情况下
针对进化群体的分析通常依靠短读长的池测序,虽然这样能够很好的得到等位基因频率的变化,但是往往忽略了其在单倍体水平的变化。本文针对重复时间序列的池测序数据,提出了一种单倍体重建的方法。该方法成功识别出了一段6.93Mb大小的单倍体区块,这一区块对果蝇的热适应有重要作用。进化重测序得到越来越广泛的应用,从病毒到细菌,从酵母再到跟高级的多细胞生物。在微生物的进化实验中,进化多起源于新的突变;对于多细胞生物,群体数量较小,进化多起源于现有变异。既往针对真核生物的进化重测序实验报告了很多很多的受选择位点,当然,其中很多是因为连锁不平衡造成的。因而要识别出真正受选择的位点,就要努力降低连锁不平衡对实验结果造成的影响。通常的做法包括:在起始种群中纳入更多类型的单倍体,使用更大的群体,经历更多的实验世代数,增加实验的重复数量。隔代施加选择(一代又选择,下一代去除选择)也能降低连锁不平衡造成的影响。
特别是当一个受选择位点位于起始种群的一个单倍体上的时候,进化重测序实验中所经历的世代数通常不足以支持足够的重组发生,因而会出现很长的单倍体区块,其中很多中性位点也因此出现受到选择的假象。此外,我们是从起始群体中抽样测序,如果抽样个体的单倍体区块和未抽样个体的单倍体区块是同一起源(即IBD),同时在未抽样个体的单倍体区块外有受选择位点,而抽样个体没有该受选择位点,这就会造成我们识别出了一个错误的单倍体区块,这个区块上并没有我们所想要的受选择位点(下图)。当然,避免这种情况出现要依靠对全部起始种群测序,而这通常很难实现。
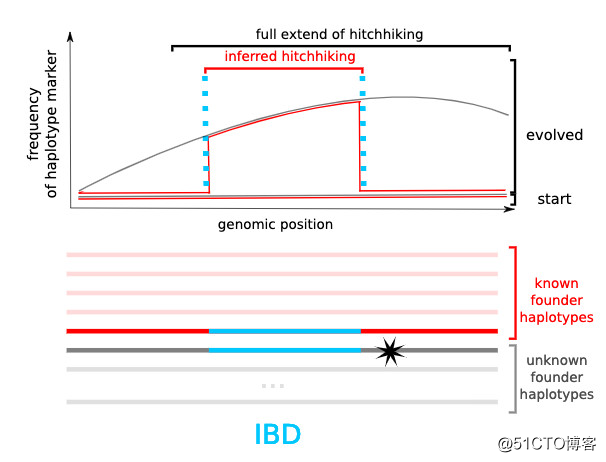
【起始群体中,我们只对红色单倍体型进行抽样测序,而受选择位点(黑色型号)位于未被抽样的起始种群,那么我们依据起始种群建立的单倍体区块上有可能不会包括真正的受选择位点。】
Franssen将识别出的SNP比对到起始群体的染色体上,发现很多SNP的频率变化出现了高度的一致性,这就提示了很多SNP是通过搭便车效应增高频率的,并且形成了一个单倍体区块。本研究提供了一种方法,在不需要对起始种群每个个体进行测序的情况下,利用搭便车SNP位点频率变化,能够精确的重建单倍体区块。
单倍体区块重建的准确度取决于整个实验过程中SNP位点的频率变化程度。变化越大,信噪比越高,准确度也越高。在重建过程中,面对大量的SNP位点,会优先考虑低频SNP,因为低频SNP更具有单倍体特异性,同时采用扫描窗口的方法,一般重组率越高,窗口越小。具体方法参见R包“haploReconstruct”。
首先在5个重复中,模拟了受选择位点的变化,两个位点相距1Mb。经过60代的进化后,受选择位点的频率显著升高。
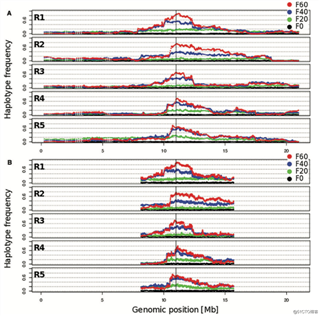
【受选择单倍体位点的可视化和重建。针对果蝇2R染色体的5个实验重复(R1-R5)。A:受选择位点的频率在60代的进化中真实的变化情况;B:通过标记重建的单倍体区块变化情况。竖直线表示受选择位点所在的位置。】
使用haploReconstruct,我们成功的重建了一段7.57Mb包含1347个标记位点的单倍体区块。而且中间结果非常好。
通过对不同实验参数组合的模拟,发现起始频率越低、扫描窗口越小、测序深度越高、标记越多、世代节点越多,单倍体重建的准确性越高。当然,实验重复数量也是一个重要影响因素,如果重复数量超过3个,那么上述的因素对结果的影响并不会太大。通过模拟结果还发现,如果将区块内最小相关性系数设在0.7,最小频率变化设在0.2,以此进行单倍体区块重建,其错误率几乎可以忽略不计。因而,本方法中,上述两个参数的默认值即为0.7和0.2。
同时还发现,重建单倍体区块的长度和准确性是一对矛盾,一些提升重建准确性的设定往往会降低重建单倍体区块的长度。而增加世代节点数不仅能够提升准确性,还可以在一定程度上增加了重建单倍体区块的长度。
将上述的方法付诸实践,应用到黑腹果蝇进化实验中。实际使用会比计算机模拟的情况更加复杂。通过使用不同的参数组合,发现识别出的受选择单倍体区块的差别不大,仅仅在区块的长度上可能存在略微不同。通过与之前研究结果的比较,可以发现这种方法有较高的准确性,成功建立了单倍体区块,其错误率在0.46%-0.87%之间。
既往很多进化重测序的研究假设SNP之间是相互独立的。但实际上并非如此,如果我们能够将位点之间的关联性考虑在我们的结果中,那么会大大提升识别选择靶标的能力。本研究中使用的方法,通过重建单倍体区块,能够准确的识别出染色体中受选择的区域。
但是本研究最大的一个缺点在于,实验方法对受选择的低频位点敏感性很高,而对于中频和高频受选择位点的识别能力有限。而且,中频位点在快速适应中往往会发挥更大的作用。这也这一定程度上影响了该方法的使用范围。
1)数据格式
使用R包haploReconstruct进行单倍体区块重建,其读入数据的格式是sync格式,sync格式是PoPoolation数据分析软件输出的一种格式,格式形式如下:
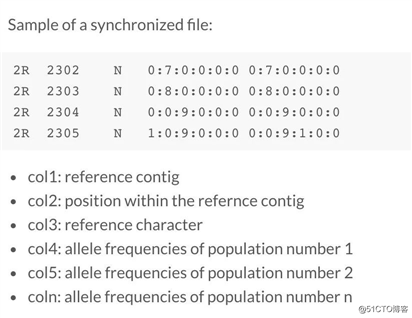
其中第4-n列为群体中A:T:C:G:N:del的频数。
参考:
https://sourceforge.net/p/popoolation2/wiki/Tutorial/
2)数据清洗
选择出含有最大信息量的SNP,包括限制minor allele的最大频率和最小频率变化等指标。
3)滑动窗口,计算窗口内SNP位点的相关性指标(Pearson相关系数)。以窗口大小的一半为步长滑动窗口。
4)确定单倍体区块
将相邻阳性窗口合并,判定为一个单倍体区块。
我们有4个重复,经过了100代的实验室进化过程,每10代进行一次抽样测序,即有11个时间节点。下面对测序染色体chr_785进行单倍体区块的重建。
library(haploReconstruct)
# designate the founding population
base_pops <- rep(c(TRUE, rep(FALSE,10)), times = 4)
# read sync file
ex_dat <- sync_to_frequencies("chr_785.sync", base.pops = base_pops, header = T)
dat_filtered = initialize_SNP_time_series(chr=ex_dat$chr, pos=ex_dat$pos, base.freq=ex_dat$basePops,
lib.freqs=ex_dat[,7:ncol(ex_dat), with=FALSE],pop.ident=rep(1:4,each=11),
pop.generation=rep(c(0,10,20,30,40,50,60,70,81,90,100),times = 4), use.libs=rep(TRUE,44))
# Reconstruct
dat_reconst = reconstruct_hb(dat_filtered, chrom=levels(ex_dat$chr))
# plot reconstructed blocks
par(mfrow = c(1,1))
plot(dat_reconst, indicate_shared=TRUE, addPoints=TRUE)
# plot the second block trajectory
par(mfrow=c(4,1),mar=c(4,4,1,1),oma=c(0,0,0,0))
tp = c(0,10,20,30,40,50,60,70,81,90,100)
plot_hbr_freq(dat_reconst, hbr_id=2, replicate=1, timepoint=tp, window=1)
plot_hbr_freq(dat_reconst, hbr_id=2, replicate=2, timepoint=tp, window=1)
plot_hbr_freq(dat_reconst, hbr_id=2, replicate=3, timepoint=tp, window=1)
plot_hbr_freq(dat_reconst, hbr_id=2, replicate=4, timepoint=tp, window=1)
# plot SNP trajectories in the second block
plot_marker_trajectories(dat_reconst, hbr_id=2)通过上述代码对chr_785染色体染色体重建,得到了3个单倍体区块,分布在该染色体的18Mb-21Mb之间。如下图:
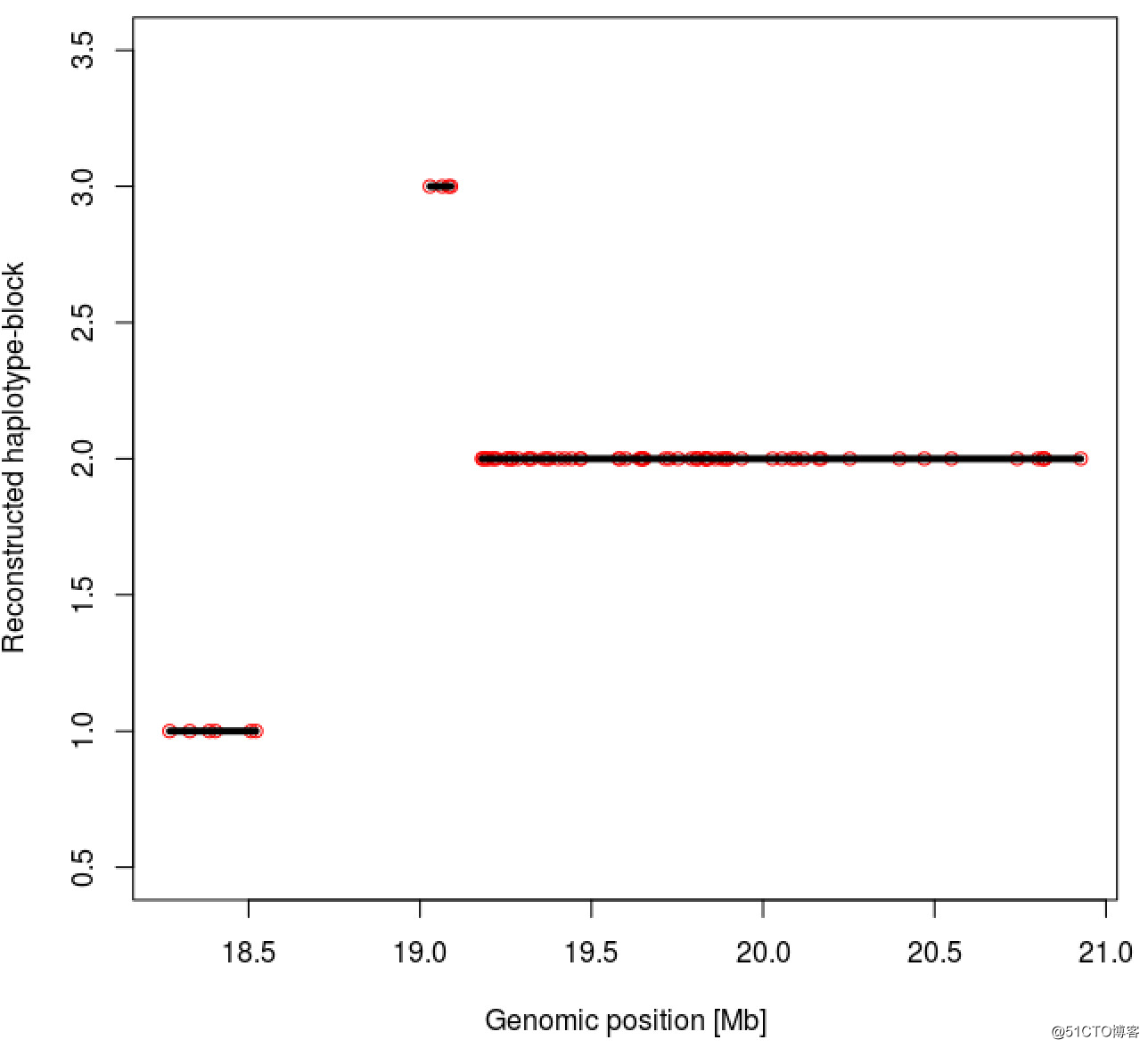
其中2号单倍体区块最长。下图是针对4个重复组的2号单倍体区块的的动态变化。
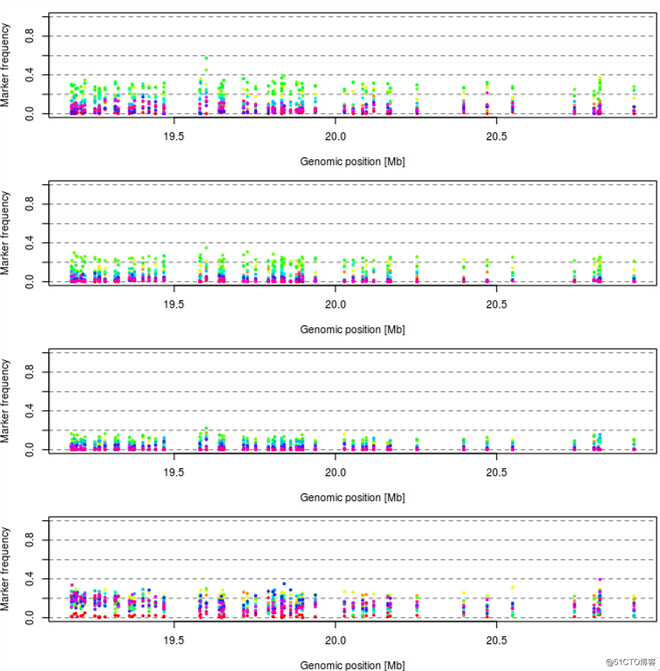
下图表示2号单倍体区块上的SNP位点随时间的动态变化。
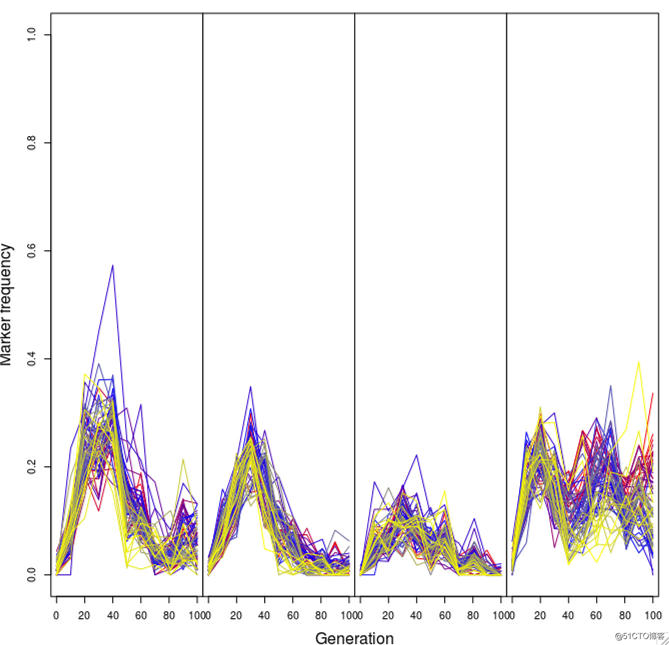
可以看出,在该区块上SNP的动态变化有高度的一致性,其中前3个重复组表现出了极高的一致性变化趋势:先增高,很快降低,并接近于0。第4个重复着先增高,后呈现动态波动。
值得注意的是,第4个重复组呈现出的变化特点在很多其他的研究中有广泛存在,比如在酵母和细菌的进化重测序实验中,很多有利突变或者变异位点并不是像我们预想的那样:等位基因频率一直增高,直到在种群中固定下来。真实的情况是,有利基因频率先增高,然后到达一个平台期,在中等基因频率波动。这提示之前的变异受到了可能平衡选择。
====== THE END =====
文献来源:Franssen, S. U., Barton, N. H., & Schl?tterer, C. (2016). Reconstruction of haplotype-blocks selected during experimental evolution. Molecular biology and evolution, msw210.

标签:rar 之间 关联性 als 关联 包括 syn 输出 情况下
原文地址:https://blog.51cto.com/15069450/2577306