标签:示意图 数值 height 意图 结构 com 图形化 一个人 聚类
8种特殊建库测序
1. RNA-seq
2. 外显子测序
3. small RNA-seq
4. 单细胞DNA测序
5. 单细胞mRNA测序
6. 甲基化测序
7.Moleculo长测序
8. Ribozero和方向性RNA文库
1. RNA-seq
今天呐,主要是给大家介绍一下RNA-seq(RNAsequencing)。也就是RNA的高通量测序技术。
因为我们这个节目主要是针对医学方面的技术进展,所以,我们今天对于RNA的介绍,也会侧重于人的RNA测序方面的技术介绍,以及相关的生物信息学方面的工作。
RNA高通量测序(RNA-sequencing,缩写为RNA-seq)是目前高通量测序技术中被用得最广的
一种技术,RNA-seq可以帮助我们了解:各种比较条件下,所有基因的表达情况的差异。
它可以检测的差异有:正常组织和肿瘤组织的之间的差异;它呐,也可以检测药物治疗前后,基因表达的差异;它呐还可以检测发育过程中,不同的发育阶段,不同的组织之间的基因表达差异。诸如此类呐,很多。我们不一一列举。
那么在所有检测的差异类型中,最常见的,就是检测所有mRNA的表达量的差异,这是最常用的一种检测。
同时呐,我们还可以检测 RNA 的结构上的差异。例如:mRNA的剪接方式的差异,也就是我们一般说的“可变剪接”,还可以检测“融合基因”,同时还可以检测基因单点突变导致的SNP(Single Nucleotide Polymorphisom)。
接下来,我们分成“RNA-seq测序方法”和“RNA-seq测序数据分析”两个部分,分别介绍RNA-seq。
RNA测序方法
在测mRNA的过程当中,首先要解决的问题,是如何去除核糖体RNA也就是去除“rRNA”(Ribosomal RNA)。
那在通常抽提到的总RNA中,绝大部分都是核糖体RNA(rRNA)。以人类的细胞或组织为例,一般抽提到的总RNA当中,95%都是核糖体RNA。剩下的2%到3%是mRNA。还有呐,2%到3%是Long non-coding RNA、或者tRNA、microRNA,这些RNA,也就是说mRNA只占了所有RNA中的一小部分。
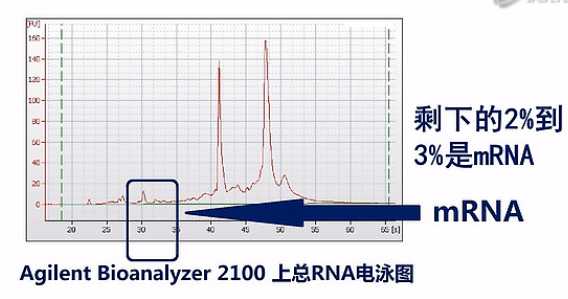
如果我们把所有的RNA都拿来测序的话呐,测到的绝大部分的序列数据呐,都是核糖体RNA。而且这当中(rRNA)比例会高达95%左右,但是呐,核糖体RNA在整个人类当中都是非常保守的,而且在人的各个组织、器官当中也是极度稳定的。也就是说,测rRNA,它得到的数据,并不能为我们实验者提供什么有用的信息,而mRNA才是RNA当中信息含量最丰富的那个部分。
我们一般的RNA-seq要测的,也是mRNA的各种变化,所以呐,在实验过程当中,我们一般要把核糖体RNA先去掉。然后再进行建库测序。
去除核糖体RNA,并进行建库的方法,有许多种。
今天呐,我们主要介绍一下应用最广泛的illumina公司的TruseqRNA建库方法。其它的方法呐,以后我们再找机会再给大家做介绍。
那么这张呐,就是mRNA测序的建库过程图。首先呐,是利用高等生物的mRNA都有Poly(A)尾巴这个特点,用带有Poly(T)探针的磁珠与总RNA进行杂交。然后呐,Poly(T)探针呐,就和带Poly(A)尾巴的mRNA结合在一起,接下来呐就回收磁珠,然后呐把这些带Poly(A)的mRNA从磁珠上洗脱下来。
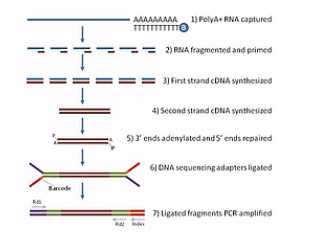

然后呐,再把这些洗脱下来的mRNA用镁离子溶液进行处理。镁离子溶液会把mRNA打断。

被打断的这些mRNA片段,再用随机引物进行逆转录。

逆转录成(第一链)cDNA后,再合成出第二链(cDNA)。这样就成为双链的cDNA。接下来呐,我们再在双链的cDNA的两端接上“Y”型的接头。这样呐,就成了标准的测序文库,然后呐,这个标准的测序文库就可以拿到HiSeq测序仪上进行测序了。



样本质量要求
在这里呐要说明一下,这个建库方法对RNA的完整度有较高的要求。也就是说,只有在mRNA大部分是完整的状态下,才能得到比较好的效果。
这是因为带Poly(T)的磁珠,它所吸附的是Poly(A)的那些序列。那么如果mRNA发生了降解,也就是mRNA断掉了,那么磁珠所吸附下来的片段,都是那些靠近3‘端的那些断片,而那些5‘端的断片呢,是吸附不下来的。会在富集过程中被洗脱掉。
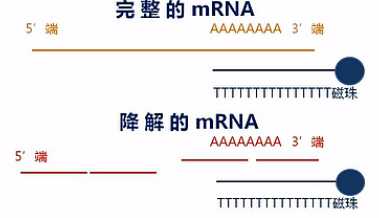
那么这样呐,接下来的数据分析当中,就会发生一定的数据偏差。
那么为了保证能够测到尽可能完整的mRNA序列呢,Illumina公司是这样建议的:它建议先对总RNA进行一次质量检测,一般是用Agilent公司出品的Bioanalyzer 2100毛细管电泳仪,对总RNA样本进行一次电泳质检。那Bioanalyzer呐会根据18S和28S这两个核糖体RNA的电泳峰是否高、是否尖,来判断RNA的质量。并且呐,会自动打分。
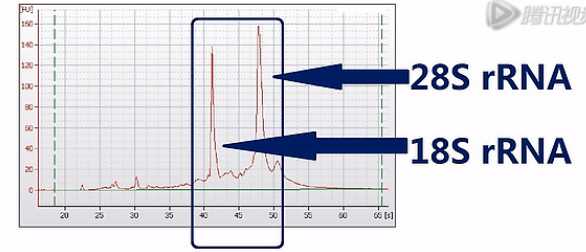
这两个峰越高、越尖,也就说明RNA的降解就越少,完整度呐就越高。那么打分呐,也会越高。反之呐,打分就会低。这个分值呐,叫“RIN”值。也就是RNA的完整度评分值。是“RNA Integrity Number”的英文首字母缩写。RIN值最高是10分,最低呐是0分。
Illumina公司推荐用RIN值在8.0以上的RNA进行建库和测序。测序完成之后呐,就可以进行数据分析了。
数据分析
分析的第一步呐,一般是先把测到的RNA片段,先mapping(比对)到基因组上,那么在比对完了之后呐,可以先看一下,有多少的RNA片段,是在靠近基因的5‘端的位置,又有多少片段在是靠近基因的3‘端的位置。
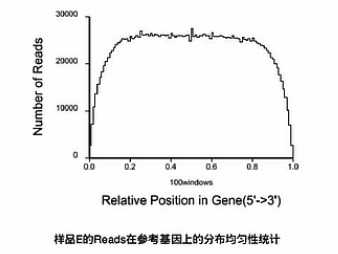
那么这张图上呐,就是把所有的基因,都按其外显子的长度呐,拉直,然后呐,归一化到“0 - 100”的这样一个长度。然后呐来看,比对上的片段,有多少是落在这0到100的这一个轴的哪个位置上。
这样一个比对的结果,就可以让我们看见前面Poly(T)磁珠在抓mRNA的时侯。
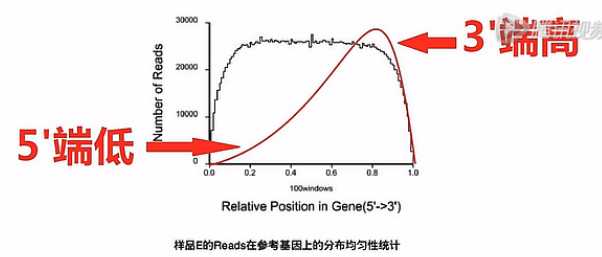
捕获下来的这些mRNA是不是完整的,如果捕获下来的这些mRNA大部分是完整的话呐,那么这个图形靠近5‘端的曲线就会显得比较饱满。它的高度会和3‘端的高度差不多。
反之,如果这根曲线的3‘端是很高的,而5‘端是比较低的,我们就可以初步判断,这个RNA有一定程度的降解。
因此,我们可以推断在捕获过程当中,有相当一部分(mRNA),它的5‘片段因为与3‘片段的Poly(A)片段的尾巴断开了,所以,没有被捕获下来。所以,这个RNA呐,是有一定程度降解的。
在知道了测序的质量之后呐,接下大家来要关注的就是不同样本之间、各个基因的mRNA的表达量的差异。
RPKM 指标
那么在做这些比较的过程当中,目前最常用的,对基因表达量进行相对定量的一个指标,就是RPKM值。那么RPKM呐,是Reads Per Kilobase of exon model perMillion mapped reads的英文的首字母缩写。
RPKM翻译成中文呐,就是每一百万条可以比对到基因组上的Read当中,有几条是可以比对到某个特定基因的,
然后呐这数值再除以该基因的外显子的长度,得到的这样一个最终的比值。

这个公式呐,就是这样的。
它的分子呐,就是经对到某个基因的外显子的read数。它的分母的第一项呐,就是这次所有比对到基因组上的read数(M reads,MillionReads)。分母的第二项,就是这个特定基因的外显子的长度。
我们接下来分步地对这个公式进行一下解释,首先呐,就是比对到某个基因的外显子上
的Read数,去除以这次所测到的、全部可以比对到基因组上的Read数。这个比较容易理解就是:这个基因所表达出来的mRNA,它所被测到的片段,来和所有被测到的、可以Mapping(比对)到基因组上的片段来进行比较。这点是比较容易理解的。
那么这个比较费解的是,为什么还要除以第二项,就是“除以这个外显子的长度”。这是因为建库过程当中,这个RNA是用镁离子溶液来处理,
然后打断(并逆录)成若干个180-200BP左右的小片段,如果一个基因的长显子越长,那么它所产生的mRNA就越长,那么mRNA越长呐,被打出来的小片段就越多。
我们来假设,一个A基因,它的mRNA的长度呐,假设它是1Kb,那么它的1Kb的mRNA可能被打成“5”个,200Bp左右的小片段;那么还有一个B基因,如果这个B基因的mRNA是2Kb长,那么,它同样被打成200Bp左右的小片段呐,它就会产生“10”个小片段。
我们来看,A基因是5个小片段,而B基因是整整10个小片段,所以,B基因在测序过程当中,它被测到的概率就会比A基因整整大出去一倍。
这就是我们为什么要把刚才第一项比出来的比值呐,然后再除以这个外显子的长度。
通过上面的解释呐,我们就可以理解:除以这个外显子的长度,它的目的:是修正这个mRNA长度所引起的mRNA的Read数的偏差。
通过这种修正呐,能够还原出一个比较真实的、原始的表达拷贝数状态。
这个呐,就是“RPKM”定义的原理。
火山图
那么作为一种针对全转录组的分析,我们希望是一次看到一个整体的样本(表达)差异的情况。而不仅仅是看少数几个基因的表达差异。
科学家做了一种叫“火山图”的一个图形,来比较形象地来说明2个样本之间的表达差异。
那么我们来看这张图,这张图呐,这个样子就象火山喷发的样子,那么这个图呐,是2个样本的RNA的表达量的对比。
这个图的横轴呐,是表示某个基因的表达是上升了,还是下降了。
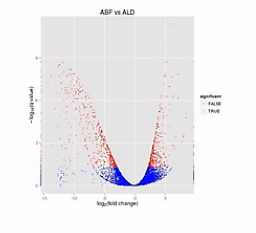
纵轴是表示这种差异的置信程度,这其中的每个点呐,就是两个样本当中同一个基因的mRNA表达量的变化。
如果这个基因的表达是上调了,那么这个点呐,就往右移动。反之,如果这个基因的表达量是下调了,那么这个点呐,就往原点的左移动。
那么这个纵轴呐,就是这种变化差异的置信程度。如果这个置信程度越高呐,那么这个点的纵轴位置也越高。
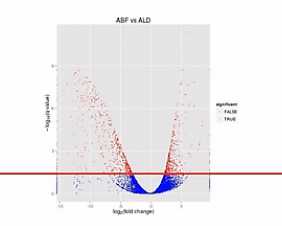
那么我们在纵轴上划了这样一条水平线,超过这个水平线以上的(点)呐,(其差异水平的)置信程度是很高的。我们就把它标示成红颜色。如果低于(这条水平线的)置信程度呐,它的置信程度也相对低一些,我们把它标成蓝颜色。
这里要解释一下,为什么差异程度是相同的情况下,它们的差异置信程度是不一样的。比如说同样是差了2的5次方,也就是32倍,它的差异置信程度会不一样,有些是蓝点,有些是红点。
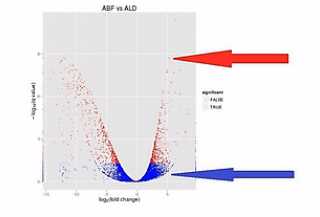
A基因在甲样本中,被测到了3200条,而在乙样本中被测到了100条;B基因在甲样本中,被测到了320条,而在乙样本中被测到了10条。它们同样是差了31倍,但是因为A基因的样本统计数,远大于B基因的样本统计数,也就是说,它们的Reads数有那么大的差距。
所以,A基因的这个差异的置信程度,会比B基因的这个差异置信程度要高许多。
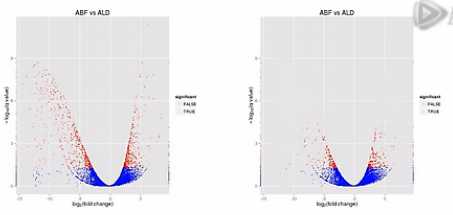
那么,我们再来对比这两张图。那么就可以比较直观地发觉,左侧的这个图当中,有更多的基因表现出明显的差异,这样呐,火山图就为我们提供了一个形象的、直观的、整体表达差异信息。
聚类分析图
聚类分析呐,是RNA分析中非常常用的一个手段。它呐是通过多个样本的全基因表达谱对比,
来找到它们之间的相似性,和相近关系。
这是一张聚类分析的图,横轴呐是样本,纵轴呐是基因。通过聚类分析呐,可以发现:在这个群体中,样本被分成了3个群体。

每个群体的内部呐,都有着相似的表达特征。同时,我们还可以看到,基因的表达,也是成簇的,这儿呐大体上分成3个基因群。那么这3个基因群呐,各自有着相似的表达量。
聚类分析呐,有很多的应用,比如说:我们可以分析疾病的亚型。
那么还可以通过对多个基因在特定疾病当中的表达倾向性呐,来找出可能的、新的、诊断用的Biomark。
聚类分析,有很多新的应用,有待我们一一去开发。
GO分析
GO分析是RNA-seq分析中非常常用的一种分析。GO是Gene Ontology的缩写,GeneOntology呐是一个国际化的、基因功能分类体系。这个体系用一整套动态更新的标准词汇、和严格定义的概念,来全面地概括任何生物中基因和基因产物的属性。
GO主要描述基因的三个属性:
第一,是这个基因,它参与的生物过程
第二,是这个基因的产物的功能
第三、是这个基因产物在细胞器内的空间定位
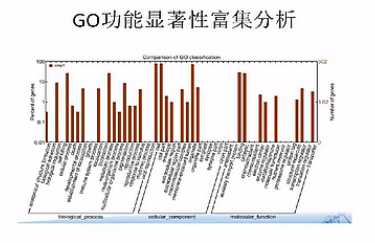
差异基因GO富集柱状图:可以直观的反映出在生物过程、细胞组分、和分子功能富集的差异基因的个数分布情况。
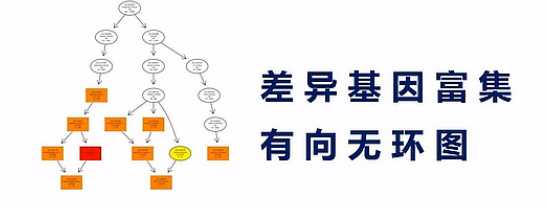
有向无环图,是差异基因GO富集分析的图形化展示方式,从上到下呐,它所定义的功能范围越来越小、越来越精准。
它的分支呐,表示包含关系。而这个圈圈的颜色越深呐,表示这个富集关系程度越高。
Pathway分析
通路分析:通路(Pathway)是指在系统水平上完成生物的某一功能的基本单元、或者局部子网络。
KEGG,也就是:Kyoto Encyclopaedia of Genes andGenomes。翻成中文:就是《京都基因和基因组百科全书》,是目前公认的、最权威的基因功能数据库。
这其中的Pathway(通路)是KEGG的核心内容。
目前针对Pathway的分析、注释,大多数是基于KEGGPathway来做的。
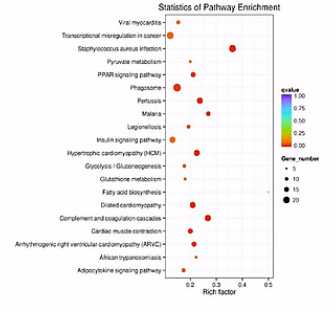
散点图是KEGG富集分析结果的图形化展示方式。
在此图中,KEGG富集程度通过Rich factor、Qvalue和富集到此通路上的基因个数来衡量。
点的面积越大呐,则富集的基因数越多。
富集的因子越大呐,则表示富集的程度越大。
qValue呐,是校正之后的pValue。那么它越接近于0呐,表示富集程度越显著。
结构变异分析
前面,我们讲的都是基于RNA表达量的差异分析。
接下来呐,我们要说一下,RNA-seq当中,可以测到的mRNA上的各种结构上的变异。
所谓结构上的变异呐,也就是RNA序列的变异。
主要呐,是3种:
第1种,是可变剪接
第2种呐,是融合基因
第3种呐,是点突变,也就是SNP
结构分析需要较深的测序深度
这里要说明一下,对于想要测mRNA结构变异的用户呢,建议测序深度要测比较深。
我们一般呐是建议测10G以上的数据量。那么为什么要测这么多的数据量呐?原因是二代测序,目前的测长还不是很长,每一个Read呐,只有大约100到125个Bp左右。如果测序深度不够呐,那么读到的这些read在整个的mRNA上的分布呐,是一种比较零碎的一种状态。
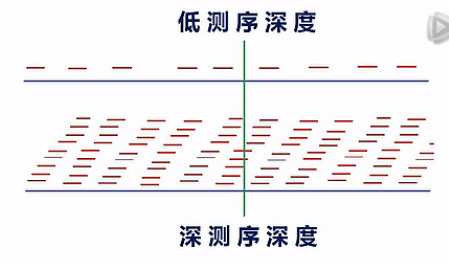
那么在这种比较零碎的、不完整的覆盖情况下,要去分析哪里有一个剪接点,哪里有一个断点,哪里有一个SNP,它不是很准确的。
当测序深度足够深的时侯,在每一个位点,都有10几次、或者几10次的覆盖的时侯呐,我们就可以比较有把握地来判断出,哪儿有了一个新的剪接点,哪儿出现了一个断点,哪儿,碱基发生了突变。
可变剪接
可变剪接,在真核生物中普通存在。一般一个人的组织样本当中呐,可以通过高通量测序,发现有5000个到20000个左右的可变剪接。
融合基因
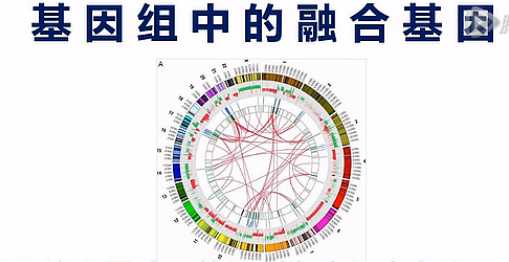
融合基因呐,是指原来在基因组上分开的2个基因,因为某种原因,染色体发生了重排。
重排的结果呐,是让A基因的头,接到了B基因的身体上,这样就产生了融合基因。
那么这张图呐,就是一个癌细胞中的融合基因的示意图。
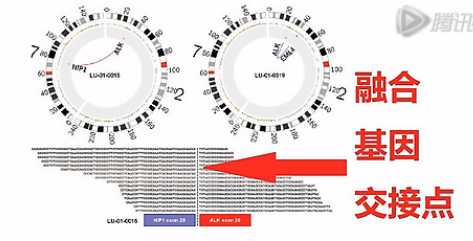
接下来这张图呐,是高通量测序测到融合基因的这个图。我们可以看到这10几个Reads都横跨在这个融合基因的、交接点的两侧,由此呐,证明了这个癌细胞当中有这么一个融合基因。
点突变
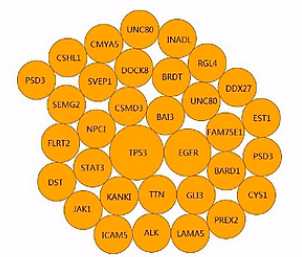
RNA-seq呐,还可以找出点突变,这个呐,是一张泡泡图,来表示我们所找到的点突变。
发生突变频率最高的这个基因,就用最大的泡泡来表示。(突变)频率低一点的,就画一个小一点的泡泡(频率),再小一点,那么再小一点的泡泡。
这些泡泡呈逆时针排列,形成这样一个泡泡图。
2. 外显子测序
3. small RNA-seq
4. 单细胞DNA测序
5. 单细胞mRNA测序
6. 甲基化测序
7.Moleculo长测序
8. Ribozero和方向性RNA文库
标签:示意图 数值 height 意图 结构 com 图形化 一个人 聚类
原文地址:http://www.cnblogs.com/think-and-do/p/6613719.html